The Reductionist Approach

The hERG assay has been a mainstay of safety screening in drug discovery for the last 2 decades. Aided by the development of high throughput automated technology, this important assay has brought unparalleled progress in our ability to design safer drugs without those properties that may lead to life-threatening cardiac arrhythmia.
In a series of posts, Dr Michael Morton explains the background to the introduction of hERG screening and why this seemingly successful approach is about to change dramatically.
Background
A number of effective and widely used drugs, across diverse chemical and therapeutic classes, have been withdrawn from the market or have had restrictions placed on their use, due to an association with a distinctive and potentially fatal cardiac arrhythmia known as Torsades de Pointes (twisting of the peaks) – see image below.
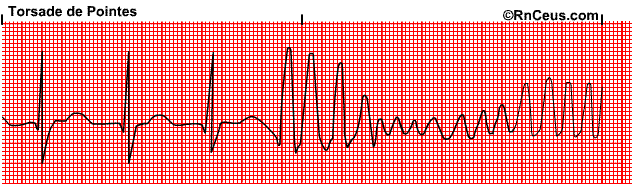
These drugs included several very successful medicines such as quinidine (an anti-malarial), cisapride (a laxative), terfenadine (an anti-histamine for allergies), clobutinol (a cough suppressant), dofetilide (an anti-arrhythmic), sparfloxacin and grepafloxacin (antibiotics). A common feature of these drugs was their ability to prolong the QT interval of the ECG, indicating a delay in cardiac repolarisation. Further investigations showed they inhibited a specific ion channel known as hERG. These channels conduct a potassium current (IKr) that is crucial for cellular repolarisation following an action potential. We now recognise there is an association between reduction in IKr (by hERG inhibition) leading to a delay in ventricular repolarisation (QT prolongation) and the potential to cause Torsades de Pointes (TdP) in some patients.
Regulatory guidance was introduced in the form of ICH guidelines (S7B and E14) requiring all new drugs to undergo a careful assessment of their potential to inhibit hERG and prolong the QT interval, both preclinically and clinically. Unfortunately, chemicals from many different series and chemotypes possess the potential to bind to, and affect conductance through, hERG channels. The pharmaceutical industry has since made huge strides in the screening out of compounds with potent hERG activity. This has been facilitated by the development of high quality, high throughput, automated electrophysiology technology that enables the rapid screening of large numbers of compounds at low cost, allowing functional data to be generated quickly enough to meet the medicinal chemist’s design-make-test cycle timings.
The reductionist approach
Assessing hERG binding or hERG potency has become an integral part of virtually all small molecule screening cascades and has led to fewer compounds that affect the QT interval entering clinical development. This principle of using activity at a single molecular target as a surrogate marker for a complex, multifactorial problem in a very small number of patients, is what may be described as a reductionist approach to the problem. Indeed, as a result of this approach, there have been no recent withdrawals from the market due to TdP.
This reductionist approach is common in drug discovery: to identify a single protein target considered important in a disease process and then seek to modulate or inhibit its activity. This has been done to great effect in search of novel medicines. However, toxicities are typically complex multifactorial processes, rarely attributable to a single causative mechanism.
Risk/benefit
In many cases, it isn’t possible to eliminate hERG activity entirely, whilst retaining other attractive features of a molecule. Therefore, a balance needs to be found between desired properties such as potency, solubility, bioavailability with a level of hERG activity that is considered low enough.
But how low does hERG potency need to be to avoid these clinical events? In making these judgements considerations include the margin between drug concentrations affecting hERG and those needed for efficacious activity, how much confidence there is in those predictions, as well as the risk:benefit in each indication. For example, compounds with hERG activity that cause a degree of QT prolongation, have been acceptable in patients with advanced cancer or severe infection. Conversely, any degree of QT prolongation in non-life-threatening indications such as asthma or arthritis has generally meant a stop for that compound. Given the costs involved in conducting clinical trials, and the late stage at which small changes in QT would be identified, there is often an abundance of caution and early de-selection of compounds with even modest hERG activity.
The issue
Today there is a recognition that not all compounds that can inhibit hERG will necessarily prolong the QT interval in patients. Furthermore, not all QT prolonging drugs predispose to TdP. Many other factors are known to be involved in the precipitation of this low incidence arrhythmia including underlying physiology, disease state, hypokalaemia or hypomagnesaemia, pharmacology and genetic predisposition. Although the reductionist approach has certainly been successful in reducing the number of drugs withdrawn for TdP, there is a growing realisation that this may have been achieved at a cost. Have we, by relying on a surrogate marker of a surrogate marker, inadvertently rejected large numbers of potentially safe and effective drugs that were not in fact, pro-arrhythmic?
The future
Given society’s large areas of unmet medical need, we must develop our scientific approaches to better identify those compounds that present a clear risk. The CiPA initiative (Comprehensive In vitro Proarrhythmia Assessment), has brought together interested parties from industry academia and regulatory authorities to address this need. In our next post, Dr Morton will discuss the advent of a new era in cardiac ion channel safety testing under CiPA that is intended to provide scientists with a more sophisticated evaluation of a compound’s pro-arrhythmic potential.




